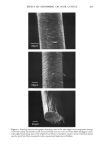
170 JOURNAL OF THE SOCIETY OF COSMETIC CHEMISTS proteins of this layer are collagen and elastin accounting for approximately 70% and 4% of the dry weight, respectively. Collagen fibers run throughout the skin and are made up of smaller tropocollagen molecules which form hydrogen bonded helices (3). In marked contrast, elastin contains many amorphous regions. About 90% of the amino acids have nonpolar side chains which stabilize the molecule by hydrophobic interactions (4). Furthermore, individual elastin fibers are covalently cross-linked forming a three dimensional network. The dermal protein fibers are embedded in a gel matrix composed of mucopolysac- charides and water, often called the ground substance. The mucopolysaccharides are high molecular weight chains of carbohydrates with covalently attached protein side chains. These macromolecules carry a large negative charge at neutral pH. Due to electrostatic repulsions along the polymer backbone, these molecules probably exist in an extended, stiff conformation under physiological condition (5,6). The elastic (time-independent) properties of skin have been the subject of many investigations (see reference 1 for a review). The results of these experiments indicate that at low strains (20%), the stress varies linearly with strain giving an elastic modulus similar in value to that obtained for pure elastin (approximately 106 dyne/cm2). At high strains, linear behavior is again observed, however, with an elastic modulus several orders of magnitude larger and very similar in magnitude to that obtained for pure collagen. Thus, elastin appears responsible for elastic behavior of skin at low strains while collagen gives skin high tensile strength at large extensions (1). Dermal protein fibers are easily visible by light microscopy. Upon microscopic inspection the fibers exhibit a regular, wavy pattern. The results of microscopy and x-ray diffraction studies suggest that collagen fibers are folded periodically producing the wavy pattern. Increasing strain serves to align the collagen and hence, the waviness vanishes (7). Several authors have suggested similar structural models for skin consistent with the observed properties (7,8). This model suggests that collagen and elastin are cross-linked at regular intervals. Tension exerted by elastin causes the collagen fibers to fold in a repeated manner. Low levels of stretching extends the elastin and aligns the collagen, while at high strain levels both the collagen and elastin molecules become extended. The time-dependent, viscoelastic properties of skin have not been extensively investi- gated. Early indications of the viscoelastic nature of skin were obtained from the results of in vitro stress relaxation experiments of Kenedi et al. (9) showing relaxation time constants over the range of 0.29 to 1700 sec. Lanir and Fung (10) used stress relaxation and hysteresis techniques to investigate directional differences in time-dependent properties of rabbit skin at large strain. Their results showed that the viscoelastic properties varied markedly along orthogonal directions of stretching. Since collagen fibers exhibit preferential alignment, the authors suggested that the large-strain, time-dependent properties were associated with the stretching of collagen fibers. More recently Finlay (11) investigated the torsional properties of skin at low strain, by measuring the phase lag between the periodic displacement and resultant torque over a range of frequencies. His results showed that, over the range of 0.04 to 1 Hz, the phase angle was independent of frequency, a viscoelastic behavior similar to that of mucøpølysaccharide-water gels. Thus, Finlay concluded that the ground substance matrix was responsible for the viscoelastic properties of skin.

MECHANICAL SPECTRA OF SKIN 171 Potts and Breuer (12) have recently investigated the low-strain stress relaxation of excised hamster skin and observed three characteristic relaxation processes, with time constants of approximately 2, 50 and 350 secs. For each relaxation, the intensity (i.e., the magnitude of the relaxation modulus measured at the peak height), but not the time constant, varied with relative humidity. It is the purpose of this investigation to study the thermal and relative humidity dependencies of these three relaxation processes, in order to gain insight into the macromolecular processes responsible for each of them. METHODS The experimental techniques have been published in detail elsewhere (12). For the purposes of this report only a brief summary of methods will be given. SAMPLE PREPARATION Adult (age 6 to 8 months) male and female hamsters were sacrificed immediately prior to experimentation. The animals were shaved and the skin removed from the back. A strip of skin 10 by 0.5 cm was cut parallel to the spine. The sample was immediately mounted inside a controlled atmosphere chamber on a tensile testing device (Instron © ) and equilibrated several hours at zero-strain under the appropriate conditions of temperature and relative humidity. This procedure was necessary in order to obtain reproducible results. STRESS RELAXATION The sample was stretched to constant percent elongation (0.6%), in a constant period of time (0.6 sec). Force data were measured over a time period of 2000 sec. at intervals of increasing duration. The data were collected and stored on a floppy disk for subsequent computer analysis. DATA REDUCTION The relaxation spectrum can be approximated as the derivative of force with respect to the logarithm of time (13). --d F(t) H(r) - -- (1) dlnt where H(r) is the relaxation modulus, F(t) is the force per unit cross-sectional area, t is the time and ß is the relaxation time constant. The relaxation data were analyzed by first smoothing the raw data and then performing an analytical differentiation (12). The relaxation modulus [H(r)] was then plotted vs logarithmic time to yield a relaxation spectrum. At each condition of temperature and relative humidity at least three separate spectra were obtained. In each spectrum the values of H(r) and LOG•0 ß associated with each peak varied by no more than plus or minus 15% and plus or minus 0.15, respectively. These spectra were then combined in a point-by-point average to yield the final spectrum. The intensity and time constant of each relaxation peak were then obtained by inspection of the final spectrum.
Purchased for the exclusive use of nofirst nolast (unknown) From: SCC Media Library & Resource Center (library.scconline.org)